Data analysis was performed using MassHunter Workstation software (Agilent Technologies) and Microsoft Excel.
Phalloidin staining and quantificationTo stain F-actin in LSECs, cells grown on glass plates were fixed with 4% paraformaldehyde (PFA) and washed with PBST (0.2% Triton-X100) three times.
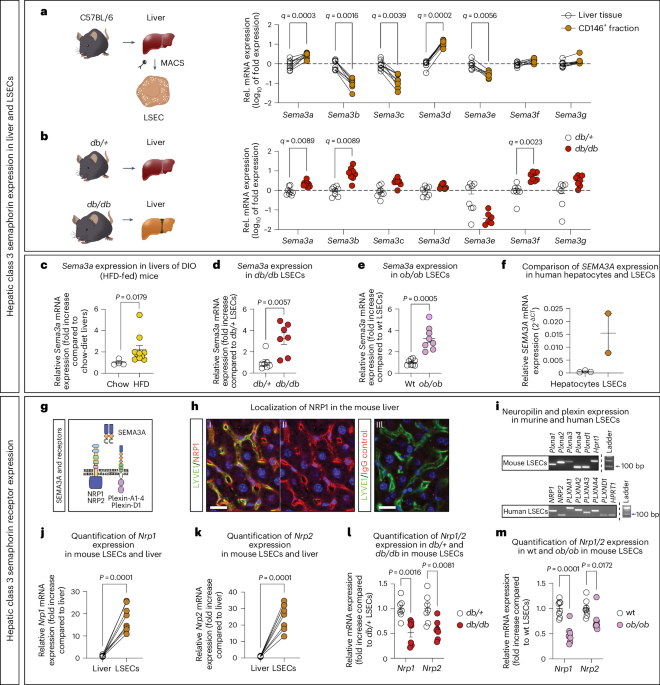
Magnetic-activated cell sortingTo isolate LSECs from the generated single-cell solution containing all hepatic cell types, the immunomagnetic cell separation system with columns from Miltenyi was used.
To count the number of fenestrae on the LSEC surface, the Cell Counter Plugin was utilized (plugins → analyze → cell counter → cell counter).
ATP measurements were performed using the CellTiter-Glo kit (G7570, Promega) according to the supplier’s instructions.
Experimental models and human donor information
Hepatocytes from various human donors were acquired from Thermo Fisher Scientific (HU4248, HU8296) and KaLy-Cell (S1426T). The donors were: female, 12 years old, white, body mass index (BMI) of 20.2, cause of death (COD) intracerebral hemorrhage-stroke (lot no. HU4248); male, 23 years old, white, BMI of 24.6, COD head trauma (lot no. HU8296); and female, 34 years old, white, BMI of 27.6, COD cholangiocarcinoma (lot no. S1426T). cDNA from these hepatocytes was obtained from elsewhere32. Human LSECs from different donors were purchased from PELOBiotech (PB-CH-153-5511). The donors were: female, 59 years old, white, BMI of 18, COD anoxia (QC-29B15F09) and male, 52 years old, white, BMI of 30.6, COD anoxia (QC-12B15F11).
Male C57BL/6J mice (Janvier), male C57BL/6N and male db/db.BKS (BKS.Cg-Dock7m +/+ LeprdbJ, JAX 000642), ob/ob.B6 (B6.Cg-Lepob/J; Jackson Laboratories, JAX 000632) and control mice were used for LSEC isolations and gene expression studies. Male heterozygous Sema3a knockout mice (C57BL/6N background54) and male wild-type littermate control mice were used to study sinusoidal porosity, hepatic lipid content and metabolic parameters and were either fed with standard chow (Sniff, V1184-300; crude protein (N × 6.25) 23%; crude fat 6.1%; crude fiber 3.3%; crude ash 6.5%; starch 34.1%; sugar 5.1%; N free extracts 49.8%; energy from fat 16 kJ%; protein 27 kJ% and carbohydrates 57 kJ%) or HFD (D12492, Research Diets, energy from fat 60 kcal%; formulation: protein (200 g casein, Latic 30 Mesh and 3 g cysteine L), carbohydrates (125 g Lodex 10 and 72.8 g sucrose); fiber (50 g Solka Floc, FCC200); fat (245 g lard and 25 g soybean oil, USP), mineral (50 g S10026B); vitamin (2 g choline bitartrate and 1 g V10001C) and dye (0.05 g blue FD&C, Alum. Lake 35–42%)) and had free access to water.
For conditional vascular EC-specific deletion of Sema3a, Cdh5-CreERT2 mice59 were mated with Sema3afl/fl (backcrossed to C57BL/6J) mice54, fed with HFD (D12492, Research Diets) for 10 weeks, injected with 75 mg kg−1 body weight of tamoxifen (Sigma, T5648) in peanut oil (Sigma, P2144) for 5 consecutive days and fed with HFD for an additional 10 weeks. Cdh5-CreERT2 mice were used as controls and were treated equally. One mouse that experienced weight loss during the final days of the experiment was excluded from subsequent analysis. For recombination analysis, DNA from liver was extracted and a genotyping PCR was performed and analyzed by agarose gel electrophoresis. The band representing the recombined Sema3a allele (delta band) was quantified by densitometric analysis using Fiji56. Genotyping was performed according to previous studies54,59. All mice were held at 22 °C (±2 °C), 55% (±5%) humidity, lighting (6:00 to 18:00). The Animal Ethics Committee of the Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen (LANUV North Rhine-Westphalia, Germany, nos. 8.87-50.10.37.09.102; 81-02.04.2022.A187, 84.02.04.2017.A305 and 81-02.04.2019.A321) and the German Diabetes Center (DDZ) Institutional Animal Welfare Committee approved all animal experiments, which were conducted in accordance with German Animal Protection Laws.
RNA isolation and RT–qPCR
To quantify gene expression in tissues or cells, mRNA was isolated using the RNeasy kit (QIAGEN). cDNA was synthesized using Oligo (dT) primers (Eurogentec) and MMLV reverse transcriptase (Promega) according to the suppliers’ instructions. qPCR was performed on a Mx3000P (Agilent Technologies) or Quantstudio 5 (Applied Biosystems) qPCR Machine using Brilliant III Sybr green (Agilent Technologies). To exclude the involvement of unspecific PCR products, –RT controls were performed and PCR melting curves of each PCR product were evaluated. Samples with faulty dissociation curves (more than two peaks) were excluded from further analysis. PCRs for all samples were run in triplicate. Relative gene expression was calculated according to Schmittgen and Livak76 using the formula 2-(C(T) gene of interest- C(T) reference gene). Finally, individual samples were plotted as fold expression with respect to the mean of the control group. For additional visualization (Fig. 1i), PCR products were separated by gel electrophoresis analysis (2% agarose gel) and documented on a ChemiDoc XRS imaging system (Bio-Rad).
Targeted lipidomics and triglyceride measurements
Diacylglycerols and ceramides were extracted from the liver and analyzed according to previous work77. Approximately 20 mg mouse liver were homogenized in 500 µl buffer cocktail (20 mM Tris-HCL, pH 7.4, 1 mM EDTA, 0.25 mM EGTA pH 7.0, 250 mM sucrose and protease and phosphatase inhibitor) using a tight-fitting glass Douncer (Wheaton). Internal standards were added to all samples. The resulting lipid phase was dried under a gentle flow of nitrogen and resuspended in methanol. For diacylglycerol and ceramide analysis, solid-phase extraction (Sep Pak Diol Cartridges; Waters) was performed. The resulting lipid phase was dried under a gentle flow of nitrogen and resuspended in methanol. The chromatographic separation of analytes was conducted using an Infinity 1290 Ultra-High Performance Liquid chromatography system (Agilent Technologies) and a reverse-phase Luna Omega C18 column, 50 × 2.1 mm, 1.6 µm (Phenomenex) operated at 50 °C. The injection volume was 1 µl. The analytes were measured as ammonium adducts (DAGs) or protonated adducts (CERs) using electrospray ionization and detected by multiple reaction monitoring on a triple quadrupole mass spectrometer (Agilent 6495; Agilent Technologies) operated in positive ion mode. Data analysis was performed using MassHunter Workstation software (Agilent Technologies) and Microsoft Excel.
Hepatic TGs were measured using a luciferase-based assay (Triglyceride-Glo Assay, Promega). In brief, approximately 25 mg mouse liver were homogenized in PBS and centrifuged. The supernatant was diluted (1:5 or 1:4) in PBS and measured as described in the manual provided.
Treatment of human LSECs and HepG2 cells
LSECs were cultured in T75 flasks coated with Speed Coating Solution (PELOBiotech, PB-LU-000-0002-00) in microvascular EC growth medium supplemented with a microvascular EC growth kit enhanced (PELOBiotech, PB-MH-100-4099). For fatty acid treatments, LSECs (passage 4–6) were passaged in 12-well dishes coated with Speed Coating Solution (PELOBiotech, PB-LU-000-0002-00) and left to attach overnight. Thereafter, different concentrations of palmitic acid (Sigma-Aldrich, P5585), sodium oleate (Sigma, O7501) diluted with fatty acid-free BSA (Sigma-Aldrich, A7039, lot SLCB3395) or fatty acid-free BSA as control were added to the cells and incubated for 2, 6, 18 and 24 h. Finally, the medium was removed and the cells were collected in 350 µl RTL lysis buffer (QIAGEN) to isolate RNA.
For treatment with FSK LSECs (passage 4–7) were seeded into coated six-well plates at 500,000 cells per well, and on the following day incubated with 100 µM FSK or dimethylsulfoxide (DMSO) for up to 6 h. The cells were collected in 350 µl RLT lysis buffer (QIAGEN) to isolate RNA and perform RT–qPCR analysis. HepG2 cells (ATCC, HB-8065) were cultured in DMEM (1×) + GlutaMax (Gibco, cat. no. 21885-025) at 37 °C with 5% CO 2 and treated in the same way as LSECs.
Flow cytometry of human LSECs
Flow cytometry was performed to determine the frequency of dead LSECs after treatment with palmitic acid or BSA. After treatment of the cells, the medium was collected and adherent cells were detached by trypsinization and transferred into FACS tubes (Falcon, 352052). FACS tubes were centrifuged (400g, 5 min) and cells were washed with PBS (Gibco, 10010-015). The centrifugation step was repeated and FVS660 (BD Biosciences, 564405, 1:1,000 dilution) diluted in PBS was added for 15 min at room temperature in the dark. Cells were washed with PBS and centrifuged for 3 min at 400g twice. The cell pellet was resuspended in PBS and FVS660+ (dead) and FVS660− (living) cells were determined using CytoFlex SRT (Beckman Coulter, CytExpert v.2.4.0.28). For quantification FlowJo software v.10 (BD Biosciences, RRIDSCR_008520) was used.
Phalloidin staining and quantification
To stain F-actin in LSECs, cells grown on glass plates were fixed with 4% paraformaldehyde (PFA) and washed with PBST (0.2% Triton-X100) three times. Then, 5 µl of stock solution (Alexa Fluor 488 Phalloidin, A12379, Abcam) was diluted with 200 µl PBS for each sample. After 30 min incubation in the dark at room temperature, plates were washed three times with PBST (0.2%) and cell nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI; Sigma Aldrich, D9542) before mounting. The staining was analyzed and imaged using a Zeiss confocal laser microscope (Zeiss LSM 710) operated by ZEN imaging software (Zen v.2.3 SP1 FP3 black). Total cell fluorescence was quantified using Fiji.
Prediction of transcription factor binding sites
The promotor sequence of the human SEMA3A sequence (−1,500 bp upstream, 500 bp downstream of the transcription start site) was loaded and analyzed using CiiiDER44 using the following parameters and databases: deficit score 0.15; JASPAR2020_CORE_vertebrates.txt; Homo_sapiens.GRCh38.94.glm; and Homo_sapiens.GRCh38.dna.primary_assembly.fa. Detected transcription factor binding sites were selected manually using the GUI interface of CiiiDER and the results were exported as an image file.
Analysis of LSEC fenestration
Liver dissociation
To generate a single-cell suspension only consisting of LSECs, the liver dissociation kit (Miltenyi Biotec, 130-105-807) from Miltenyi was utilized. First, PEB solution was prepared (47.5 ml MACS rinsing solution and 2.5 ml BSA/EDTA per animal). This mixture was de-gassed in a magnetic mixer for 15 min. Meanwhile, 500 μl coating solution (PELOBiotech, PB-LU-000-0002-00) was added into wells (24-well plate) and incubated for 30 min at room temperature. Livers were collected and transferred into a gentleMACS C-tube containing the dissociation mix from Miltenyi. The tube was closed and attached onto a sleeve of the gentleMACS Octo Dissociator after which the samples were resuspended and added onto a MACS SmartStrainer (70 μm). Then, 5 ml DMEM were added to the C-tube to collect any remaining cells and applied onto the strainer as well. Last, the Falcon tubes containing the separated cells were centrifuged at 300g for 10 min.
Magnetic-activated cell sorting
To isolate LSECs from the generated single-cell solution containing all hepatic cell types, the immunomagnetic cell separation system with columns from Miltenyi was used. The last step of the liver dissociation procedure is the centrifugation of the Falcon tubes containing the separated cells. Next, the supernatant was carefully aspirated, the pellet resuspended with 5 ml PEB and then centrifuged again at 300g for 10 min. Meanwhile, LS columns for magnetic separation were equilibrated with 3 ml PEB. After centrifugation of cells, the supernatant was removed, the pellet resuspended in 90 μl PEB and 10 μl of magnetic beads coupled to a CD146 antibody (Miltenyi Biotec; 130-092-007) were added. The Falcon tubes, containing the cell suspension and the magnetically labeled CD146 antibodies, were put onto a rotator in the fridge (4 °C) for 15 min. Afterwards, the cells were washed with 1 ml PEB and centrifuged at 300g for 10 min, then the supernatant was taken off and the pellet resuspended in 500 μl PEB. This cell suspension was applied onto a column and washed with 3 ml PEB twice. The columns were removed from the magnetic field and with a plunger, the magnetically labeled cells were washed out with 5 ml PEB onto the second column, to which a MACS SmartStrainer (30 μm) was attached. After the column and the MACS SmartStrainer were washed twice with 3 ml PEB, the magnetically labeled cells were flushed out with 5 ml PEB into a fresh 15 ml Falcon tube, which was centrifuged at 900g for 3 min. Next, the supernatant was taken off and the pellet was resuspended in pre-warmed EBM-2 medium, which resulted in 60,000 cells per well, and then incubated at 37 °C and 5% CO 2 for 4 h, after which the cells could be further utilized.
Fluorescence-activated cell sorting
For mRNA expression analyses, LSECs of 12-week-old db/db, db/+, ob/ob and wild-type control mice were isolated via MACS (see above) and additionally enriched via FACS, yielding an LSEC purity of >95%. As MACS was only necessary for pre-enrichment, cells were applied to only one MACS column but were washed three times. The magnetically labeled cells were flushed out with 4 ml PEB directly into FACS tubes and centrifuged for 5 min at 300g. Next, the supernatant was discarded and cells were resuspended in 300 µl PEB buffer containing 1:50 anti-mouse CD146 PE-conjugated antibody (Miltenyi, 130-118-253). After 15 min of incubation at 4 °C, cells were washed twice with 3 ml PEB buffer and centrifuged at 300g for 3 min. Cells were resuspended in 2 ml PEB buffer and up to 200,000 single CD146+ LSECs per mouse were sorted at a CytoFLEX SRT (Beckman Coulter).
Treatment of mouse LSECs with Semaphorin-3A-Fc
After allowing LSECs to grow for 4 h in EBM-2 medium with supplements, the cells were starved for another hour using EBM-2 medium without supplements. After 1 h, the medium was aspirated and treated with either a control protein (IgG2a-Fc, Recombinant Mouse IgG2a-Fc Protein, R&D Systems, 4460-MG-100) or different concentrations of recombinant semaphorin-3A (SEMA3A-Fc, Recombinant mouse semaphorin-3A Fc Chimera Protein, R&D Systems, 5926-S3-025) reconstituted in PBS; however, the total amount of protein was kept constant. After the cells were incubated at 37 °C and 5% CO 2 for the desired amount of time, they were fixed in either PFA (4% in PBS) or glutaraldehyde (2% in sodium cacodylate buffer).
Antibody and inhibitor treatments of LSECs
After 4 h of incubation, isolated LSECs were treated with different types of NRP1 antibodies (anti-NRP1SEMA3A; Genentech47, anti-NRP1; R&D Systems, AF566), while anti-NRP1VEGF (Genentech) served as a control47. The antibodies were diluted with EBM-2 medium without supplements (to simultaneously starve the cells) at a final concentration of 5 µg ml−1. After addition of the antibodies, the cells were incubated at 37 °C and 5% CO 2 for 1 h.
If LSECs were to be pretreated with the LIMK1 inhibitor LIMKi 3 (Tocris, 4745), they were allowed to grow 4 h and then incubated with LIMKi 3 for 1 h at 37 °C and 5% CO 2 . The inhibitor was diluted to a final concentration of 3 µM in EBM-2 medium without supplements and DMSO with a final concentration of 0.1%. As a control, the cells were treated with EBM-2 medium without supplements with the same concentration of DMSO (0.1%).
SEM of mouse LSECs
After treatment, the glass plates were removed from the wells and transferred to a 24-well plate containing 500 μl of glutaraldehyde solution (2% in sodium cacodylate buffer in a total of 2 ml: 160 μl 25% glutaraldehyde (stock) solution + 1,840 μl sodium cacodylate buffer (0.1 M)) per well. The next day, the glutaraldehyde solution was taken off and 500 μl sodium cacodylate buffer (0.1 M) was applied onto each glass plate. Following this, the sodium cacodylate buffer (0.1 M) was taken off and the cells were incubated with 500 μl OsO 4 solution (4 ml total: 3 ml 0.1 M sodium cacodylate buffer + 1 ml 4% OsO 4 ) per well for 30 min. Next, the cells were washed twice with 500 μl of cacodylate buffer for 5 min. Then, 500 μl of 70% ethanol was added into each well and incubated for 5 min. This step was repeated with 80% and 90% ethanol after which the glass plates were transferred into a 24-well plate containing 500 μl of 100% ethanol. Last, the cells were chemically dried using tetramethylsilane (TMS) (ACROS Organics, Thermo Fisher Scientific). The TMS was added into each well, approx. until the volume doubled (1:1 ratio of ethanol to TMS). After 30 min of incubation, TMS was again added until the volume doubled and incubated for 30 min. Thereafter the cells were aspirated and a few drops of TMS were added into each well, just covering the glass plate and incubated for 30 min. After the cells were aspirated, a few drops of TMS were added into each well and the plates were left to dry overnight. The plates were removed and attached onto SEM Specimen Stubs (12.5 mm Ø, 3.2 × 8 mm pin) using double-sided adhesive circles. Using a sputter coater, the plates were coated with a thin layer of gold. After this step, the samples were ready to be examined by SEM. For image acquisition, the Leo 1430 VP SEM, Zeiss FIB-SEM 540 Crossbeam or Zeiss SUPRA 55VP, together with the Zeiss imaging software, were utilized.
Manual quantification of fenestrae diameter, frequency and porosity
For the morphologic analysis of LSECs, the images obtained with the Leo 1430 VP were examined using the Fiji imaging-processing package56. Analyzed features were the fenestrae frequency (the number of fenestrae per μm2), the LSEC porosity (the ratio of fenestrated area to the analyzed cell area) and the fenestrae diameter. First, the scale was set from pixel to μm, to measure all parameters in the intended unit. Next, the cell area was determined, using the polygon selection tool. The outline of the cells was traced and the area was measured in μm2. To count the number of fenestrae on the LSEC surface, the Cell Counter Plugin was utilized (plugins → analyze → cell counter → cell counter). For a better resolution, the contrast and brightness were adjusted and the processing tool ‘smooth’ was applied (image → adjust → brightness/contrast, process → smooth). Then, the fenestrae were counted and a copy, where all the fenestrae are flagged, was saved to aid the measuring of the fenestrae diameter. The diameter was measured using the straight-line tool and the measurements were given in μm. All obtained measurements were used to calculate above-mentioned parameters by using equations (1) and (2).
$${{\mathrm{Fenestration}}\,{\mathrm{frequency}}\,({\upmu}{{\mathrm{m}}}^{-2})=\frac{{\mathrm{Number}}\,{\mathrm{of}}\,{\mathrm{fenestrae}}}{{\mathrm{Analyzed}}\,{\mathrm{cell}}\,{\mathrm{area}}\,({\upmu}{\mathrm{m}}^{2})}}$$ (1)
$${{\mathrm{LSEC}}\,{\mathrm{porosity}}=\frac{{\Sigma}\,{\mathrm{Fenestrae}}\,{\mathrm{area}}\,({\upmu}{\mathrm{m}}^{2})}{{\mathrm{Analyzed}}\,{\mathrm{cell}}\,{\mathrm{area}}\,({\upmu}{\mathrm{m}}^{2})}}$$ (2)
Quantification of fenestrae diameter, frequency and LSEC porosity with machine learning
For the morphologic analysis of LSECs, the images obtained with the Zeiss FIB-SEM 540 Crossbeam or SUPRA 55VP were examined using a deep-learning workflow that is based on the uncertainty-aware variant78 of the Contour Proposal Network (CPN)79. This model was specifically chosen for its capability to directly predict object contours in biomedical image data, providing an accurate representation of object shapes and sizes. It uses a U-Net architecture80 with a ResNeXt-101 encoder81. This setup utilized a pretrained network (ginoro_CpnResNeXt101UNet-fbe875f1a3e5ce2c) from the celldetection Python package (https://github.com/FZJ-INM1-BDA/celldetection), designed for multimodal cell segmentation. The model was fine-tuned using manual annotations and applied with an ensemble strategy. Computations were performed on the JUWELS supercomputer82.
G-actin/F-actin in vivo assay biochem kit
Quantification of F-actin and G-actin in primary mouse LSECs was performed using the G-Actin/F-Actin In Vivo Assay Biochem kit from Cytoskeleton (cat. no. BK037). To this end, LSECs were isolated using MACS, incubated for 4 h, starved for 1 h and treated for 1 h with 1 µg ml−1 of either SEMA3A-Fc or IgG2a-Fc. The division of F-actin and G-actin was performed according to the description of the kit. Afterwards, both fractions were analyzed using western blotting (antibody used was anti-actin monoclonal antibody (clone 7A8.2.1; cat. no. AAN02-S)). For quantification, a dilution series was used to generate a standard curve.
Western blotting
For the western blot sample preparation, cells were lysed with radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, pH 7.4, Sigma-Aldrich; 150 mM NaCl, Roth; 1 mM EDTA, Ambion; 1 mM Na 3 VO 4 , Sigma-Aldrich; 1 mM NaF, Sigma-Aldrich, 0.25% sodium deoxycholate, AppliChem; 1% IGEPAL, Sigma-Aldrich, in H 2 O plus protease inhibitor, Sigma, 11697498001 and phosphatase inhibitor, Sigma, 4906845001). The lysates were disrupted (Disruptor Genie, Scientific Industries) and centrifuged at 4 °C, 15,700g (Centrifuge 5415R, Eppendorf) and the supernatant was collected. Protein concentrations of the samples were determined using a Pierce BCA protein assay kit (Thermo Scientific, 23225) and all samples were diluted to the amount of the sample with the least amount of protein, while containing 20 μg at most. All samples were filled up with water to 30 μl, 10 μl 4× Laemmli sample buffer (180 μl 4× Laemmli stock, 20 μl NaF, 40 μl Protease inhibitor (Roche) and 10 μl β-mercaptoethanol) was added and the samples were incubated at 95 °C for 5 min for protein denaturation. Following, the samples were put on ice for immediate use. A Mini-PROTEAN TGX Stain-Free Protein Gel was loaded with 10–15 μl sample per lane. As a ladder 5 μl of PageRuler Prestained Protein Ladder (Thermo Fisher) was used. Gels were run at 120 V for approximately 40 min. The stain-free gel was immediately activated using UV light for 5 min. After imaging the gel, two ion transfer stacks and the blotting membrane were assembled in the transfer chamber of the Trans-Blot Turbo according to the manufacturer’s instructions. Following the transfer, the blot was imaged using Bio-Rad ChemiDoc MP Imaging software. Then, the blot was blocked in 5% milk in 1× PBST for 1 h.
To visualize cofilin-1 and p-S3-cofilin-1, the blots were incubated for at least 16 h or overnight in the primary antibody at 4 °C on a horizontal shaker (antibodies were p-S3-cofilin-1, Cell Signaling, 3313T, 1:750 dilution; cofilin-1, Cell Signaling, 5175T, 1:750 dilution and GAPDH, Abcam, ab9485, 1:2,500 dilution).
Afterwards the blots were washed three times with 1× TBST for 5 min and then incubated for 1 h with the secondary antibody (antibodies were anti-rabbit IgG, HRP-linked antibody, Jackson Immuno Research, 711-035-152, 1:4,000 dilution; and anti-rabbit IgG, HRP-linked antibody, Invitrogen, G21234, 1:2,000 dilution) on a horizontal shaker at room temperature. They were washed again three times with 1× TBST for 5 min before applying Pierce ECL Western Blotting substrate (Thermo Fisher) onto the membrane to detect specific protein bands. The membrane was incubated in the substrate for 5 min and the ChemiDoc MP and the ImageLab v.4.1 software from Bio-Rad were used to develop and analyze images.
Luminescent cell viability assay
LSECs were isolated, plated onto white, opaque 96-well cell culture plates, incubated for 4 h and fasted for 1 h, all at 37 °C and 5% CO 2 . Last, they were treated with 0, 0.5 or 2 μg ml−1 SEMA3A-Fc for 1 h, while they stayed at 37 °C and 5% CO 2 for 30 min. Then they were taken out of the incubator and equilibrated at room temperature for another 30 min. ATP measurements were performed using the CellTiter-Glo kit (G7570, Promega) according to the supplier’s instructions. Luminescence was finally measured using the Promega GloMax with the firmware v.4.88.0 and the software v.2.4.1 (emission filter, none; and integration time, 0.3 s).
Kinase activity profiling
The PamGene assays measure kinase activity in cell and tissue lysates by measuring the phosphorylation of peptide representations of kinase targets/substrates that are immobilized on the PamChip microarrays. The active kinases in the sample lysates will phosphorylate their target on the array. Generic fluorescently labeled antibodies that recognize phosphorylated residues are used to visualize the phosphorylation. We employed both types of PamChip microarrays, the PTK and the STK microarray, with 340 different substrates in total.
To perform kinase activity profiling, mouse LSECs were isolated using MACS and cells were seeded at 1 × 106 cells per well on a six-well plate. After 4 h, cells were starved for 1 h and then treated for 10 min with 1 µg ml−1 of either SEMA3A-Fc or IgG2a-Fc. Afterwards, the plate was put on ice, the culture medium was removed and cells were washed with cold PBS. After removal of PBS, the washing step was repeated. Lysis buffer (Halt Phosphatase Inhibitor Cocktail and Halt Protease Inhibitor Cocktail EDTA free, 1:50/1:100 diluted in M-PER Mammalian Extraction Buffer) was added to the cells and cells were collected using a cell scraper. Cells were lysed by pipetting up and down several times over the course of 15 min on ice. Samples were centrifuged for 15 min at 16,000g at 4 °C. The lysate was collected and transferred to a clean vial on ice. After snap-freezing in liquid nitrogen, samples were stored at −80 °C until transport to the PamGene facility. The analysis and data processing were performed by PamGene (‘s-Hertogenbosch, Netherlands).
Liver perfusion for SEM preparation
Liver perfusion and fixation were performed according to the protocol from Cogger et al.83. After fixation, the livers were placed onto specimen stubs (12.5 mm Ø, 3.2 × 8 mm pin) using double-sided adhesive circles. Using a sputter coater, the plates were coated with a thin layer of gold in a controlled and even manner. After this step, the samples were ready to be examined by SEM. For image acquisition, the Leo 1430 VP SEM, Zeiss FIB-SEM 540 Crossbeam or Zeiss Supra 55VP, together with the Zeiss imaging software, were utilized.
Semi-automatic quantification of SEM images of liver sinusoids
SEM images were quantified using Fiji with help of the trainable WEKA Segmentation plugin56,57. First, an automatic contrast (‘normalize local contrast’) was calculated for each SEM image and the polygon selection tool of Fiji was used to manually mark the area of interest (sinusoid area) and all non-sinusoid area and gaps were cleared. Next, the fenestrae area and sinusoid cell surface area was identified using a dataset-trained-classifier segmentation (WEKA) algorithm in Fiji56,57. The classifier was trained using typical SEM images and stored in a classifier file. The classifier segmentation (WEKA) algorithm led to the generation of probability maps for cell surface area and fenestrae area (Extended Data Fig. 5a). Finally, the maps were used to calculate the overall surface area of the sinusoid and also to quantify fenestrae using the ‘analyze particles’ feature, which returns the area and diameter of each object. Small objects or objects with a low circularity (circularity <0.50) were excluded from the analysis, as fenestrae are expected to be round or oval in shape. The data were transferred to Excel (Microsoft) and the frequency (no. fenestrae per area) and porosity (fenestrae area per area analyzed) was calculated.
Histology and Oil Red O staining of liver sections
Hepatic TG content was quantified by staining liver cryosections with ORO. To this end, freshly isolated livers were frozen in OCT medium (TissueTek) using dry ice. Liver cryosections (12 µm) were stained in filtered ORO working-solution (24 parts stock solution (300 mg ORO, Sigma-Aldrich, O-9755 in 100 ml 2-propanol) + 16 parts demineralized water). Sections were rinsed briefly in demineralized water and washed for 10 min in running tap water and embedded using Fluoroshield (Sigma-Aldrich, F6182). An Eclipse Ti-S microscope (Nikon) and a DS-2Mv camera operated by NIS-Elements software (Nikon) were used for imaging. Fiji56 was used to measure sections and the lipid droplet area. Finally, the ratio of stained/section area was calculated for each image. For paraffin sections, H&E and Elastica van Gieson staining was performed at the Histopathology Diagnostic Laboratory of the Institute of Pathology using an autostainer (TissueTek Prisma) according to standardized protocols. For H&E-staining of cryosections, sections were stained with Mayer’s hematoxylin (Sigma, MHS 16) and eosin Y solution (Sigma, E4282), dehydrated and embedded in Entellan (Sigma, 1.07961.0100). For PSR staining, cryosections were stained for 60 min in PSR solution (0.1 g Sirius Red, Direktrot 80; Sigma 365548 in 100 ml saturated aqueous picric acid), dehydrated and prepared for microscopy.
Immunohistochemical staining of cryosections
Immunofluorescence staining was performed with liver cryosections (12 µm) of male C57BL/6J mice that were fixed with 4% PFA. Sections were treated with blocking solution (10% normal donkey serum, 2% BSA, PBS and 0.2% Triton-X100) for 1 h and incubated overnight with primary antibodies in blocking solution using rabbit anti-LYVE1 (Abcam, AB14917, lot GR320055-2); goat anti-NRP1 (Research and Development, AF566, lot ETH0612091) or isotype control goat IgG (Santa Cruz, SC2028, lot A2913). After washing the sections in PBS (0.2% Triton-X100) for 3 × 5 min, sections were incubated for 1 h at room temperature with secondary antibodies (donkey anti-goat Alexa 555, Invitrogen, A21432, lot 1818686; donkey anti-rabbit Alexa 488, Invitrogen, A21206) and DAPI (1 µg ml−1; Sigma-Aldrich, D9542). Finally, sections were washed with PBS (0.2% Triton-X100) for 2 × 5 min and embedded using Fluoroshield medium (Sigma-Aldrich, F6182) and a coverslip. Staining was analyzed and imaged using a Zeiss confocal laser microscope (Zeiss LSM 710) operated by ZEN imaging software (Zeiss). Images were analyzed using Fiji56.
Serum parameters
To measure TGs, ALT, AST, total cholesterol (Chol) and high-density lipoprotein (HDL) cholesterol in the serum of fasting mice, Kenshin-2 Spotchem 4430 test stripes were used in combination with the SPOTCHEM EZ SP-4430. Values <15 (n.d.) were defined as 15. The samples were measured according to the manufacturer’s description. NEFA was measured using the NEFA-HR(2) Assay (FUJIFILM Wako Chemicals) and insulin was measured using an ultra-sensitive rat insulin ELISA (Crystal Chem, cat. no. 90060). HOMA-IR was calculated as (insulin × glucose (ng ml−1 × ml dl−1)/405) and Adipo-IR (FFA × insulin (mmol l−1 pmol−1) and expressed as percentage of control.
Glucose tolerance test
Mice were subjected to an overnight fast before undergoing GTTs. In the test, glucose (1 mg g−1 body weight) was intraperitoneally injected and blood glucose levels were assessed by obtaining blood samples from the tail tip. Using a Monometer Futura glucometer (MedNet), blood glucose concentrations were measured twice at each time point. To measure plasma insulin concentrations, small amounts of blood were collected from the tail tip using EDTA-coated tubes, followed by plasma preparation through a 10-min centrifugation at 2,000g. Insulin concentrations were subsequently measured using an ultra-sensitive rat insulin ELISA (Crystal Chem).
VLDL secretion assay
Mice were weighed and subjected to a 4-h fast before undergoing the VLDL secretion test. In the test, 0.5 g kg−1 body weight of WR1339 (Sigma, T8761) was intraperitoneally injected and blood was collected from the tail tip pre-injection and after 1, 2, 4 and 6 h after Triton WR1339 injection using EDTA-coated tubes, followed by plasma preparation through a 10-min centrifugation at 2,000g. TGs were measured using a LabAssay Triglyceride kit (FUJIFILM Wako Chemicals Europe) according to the suppliers’ instructions.
Metabolic cage analysis
Metabolic cages (PhenoMaster, TSE-System) were employed to measure parameters such as physical activity and food intake. Following an adaptation phase in the cages used for measurements, activity and metabolic parameters were continuously monitored. Infrared sensor frames recorded activity, and a control unit identified interruptions in the infrared sensors. Relevant data were registered by a computer using the PhenoMaster software from TSE Systems. Body weight, food and water intake, carbon dioxide production, oxygen consumption and cage temperature were quantified through integrated sensors. The respiratory exchange ratio and energy consumption were calculated by the PhenoMaster software and normalized to body weight and lean mass for relevant parameters. Lean and fat mass was measured using an NMR Analyzer (Minispec, Bruker).
Statistical analysis
All imaging analyses were performed under blinded conditions. Data were gathered and processed using Excel (Microsoft) and then transferred to GraphPad Prism (v.9.4.0) to generate all graphs. All data points were plotted individually together with the mean and s.e.m. Statistical analysis was performed using GraphPad Prism (v.9.4.0). No statistical outlier tests were applied. If necessary, samples/data were solely removed based on technical issues during the experiments. A two-tailed unequal variance t-test (Welch’s test) was used to determine statistical significance between two independent experimental groups. A pairwise Student’s t-test was performed to determine statistical significance for samples of the same mouse. In case of multiple t-tests in the same analysis in Figs. 1a,b and 2a,b, a multiple two-tailed t-test (paired or unpaired) with a two-stage step-up method according to Benjamini, Krieger and Yekutieli84 was used to correct for multiple comparisons and to detect significant discoveries. For more than two experimental groups with one or two factors, a one- or two-way analysis of variance (ANOVA) (with or without repeated measurements) was conducted, followed by a Dunnett’s, Tukey’s or Sidak’s post hoc test. Information about the performed statistical tests and samples sizes is indicated in the figure legends. Only P and q values <0.05 are shown in the figures.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.